データの可視化
最終更新日:2024-09-08 | ページの編集
概要
質問
- Rによる可視化
目的
- ggplotを使って散布図、箱ひげ図、折れ線グラフなどを作成する。
- ユニバーサルプロット設定を行う。
- ファセットとは何かを説明し、ggplotでファセットを適用する。
- 既存のggplotプロットの美学(軸ラベルや色を含む)を修正する。
- データフレーム内のデータから複雑でカスタマイズされたプロットを作成。
このエピソードは、Data Carpentriesの_Data Analysis and Visualisation in R for Ecologists_レッスンに基づいています。
データの可視化
必要なパッケージをロードすることから始める。
**ggplot2**は、 **tidyverse**パッケージに含まれています。
R
library("tidyverse")
ワークスペースにまだない場合は、前回の レッスンで保存したデータをロードします。
R
rna <- read.csv("data/rnaseq.csv")
Data Visualization Cheat Sheet](https://raw.githubusercontent.com/rstudio/cheatsheets/main/data-visualization.pdf)
は ggplot2 の基本からより高度な機能までをカバーし、
パッケージで利用可能な多くのデータ表現
の概要を理解するためのリマインダとしてだけでなく、手助けになるでしょう。
Thomas Lin Pedersen氏による以下のビデオ チュートリアル(パート1と 2)
も非常に参考になる。
ggplot2`によるプロット
ggplot2は、データフレーム内のデータから複雑な プロットを簡単に作成できるプロットパッケージである。 どの変数をプロットするか、どのように表示するか、 、一般的なビジュアル・プロパティを指定するための、よりプログラム的な 。ggplot2`を支える理論的基盤は、Grammar
of Graphics (@Wilkinson:2005)である。
この
アプローチを使用すると、基礎となるデータが変更された場合、または棒グラフから散布図に変更することを決めた場合に
、最小限の変更で済む。 これは、
、最小限の調整で出版品質のプロットを作成するのに役立ちます
、微調整。
ggplot2に関する本(@ggplot2book)があり、 。 第3版は現在準備中で、 [オンラインで自由に利用できる](https://ggplot2-book.org/)。 のggplot2`
ウェブページ(https://ggplot2.tidyverse.org)に十分なドキュメントがあります。
ggplot2の関数は、'long'形式のデータ、つまり、 すべての次元を表す列と、すべてのオブザベーションを表す行を持つ。 よく構造化されたデータ 、ggplot2`で図を作成する時間を大幅に節約できる。
ggplotグラフィックスは、新しい要素を追加することによって段階的に構築される。 この方法で レイヤーを追加すると、プロットの広範な柔軟性と カスタマイズが可能になる。
Grammar of Graphicsの背後にある考え方は、同じ3つのコンポーネントからすべての グラフを構築できるということである:(1)データセット、(2)座標系、 、(3)ジオム、つまりデータ点を表す視覚的マーク 1 である。
ggplotを構築するために、 、さまざまなタイプのプロットに使用できる以下の基本テンプレートを使用する:
ggplot(data =<DATA>, mapping = aes(<MAPPINGS>)) +<GEOM_FUNCTION>()- ggplot()
関数を使用し、data`引数を使用して特定のdata frameにプロットをバインドする。
R
ggplot(data = rna)
- (
aes)関数を使用して)マッピングを定義する。 、プロットする変数を選択し、 、x/yの位置や、 、サイズ、形、色などの特徴として、グラフでどのように表示するかを指定する。
R
ggplot(data = rna, mapping = aes(x = expression))
-
追加 ‘geoms’ - ジオメトリ、つまりプロット内の データのグラフ表現(点、線、棒)。 ggplot2`は多くの 、様々なジオムを提供している:
* 散布図やドットプロットなどには `geom_point()` を使用する。 * * `geom_boxplot()` ボックスプロット! * トレンドライン、時系列など。
プロットにgeom(etry)を追加するには + 演算子を使います。
geom_histogram() をまず使ってみよう:
R
ggplot(data = rna, mapping = aes(x = expression)) +
geom_histogram()
出力
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.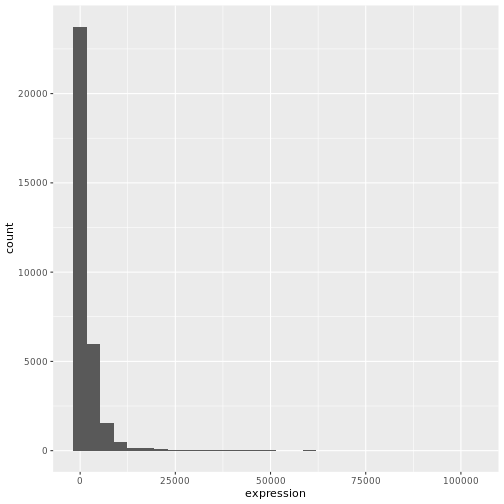
ggplot2パッケージの+は特に便利で、 、既存のggplot`オブジェクトを修正することができる。
つまり、 、プロット・テンプレートを簡単に設定し、
、さまざまなタイプのプロットを便利に調べることができる:
R
# Assign plot to a variable
rna_plot <- ggplot(data = rna,
mapping = aes(x = expression))
# Draw the plot
rna_plot + geom_histogram()
R
# change bins
ggplot(rna, aes(x = expression)) +
geom_histogram(bins = 15)
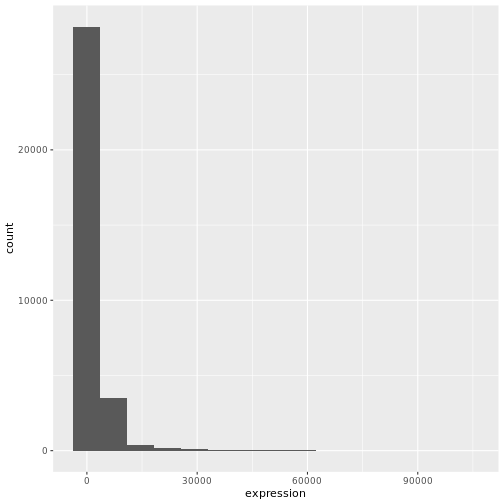
R
# change binwidth
ggplot(rna, aes(x = expression)) +
geom_histogram(binwidth = 2000)
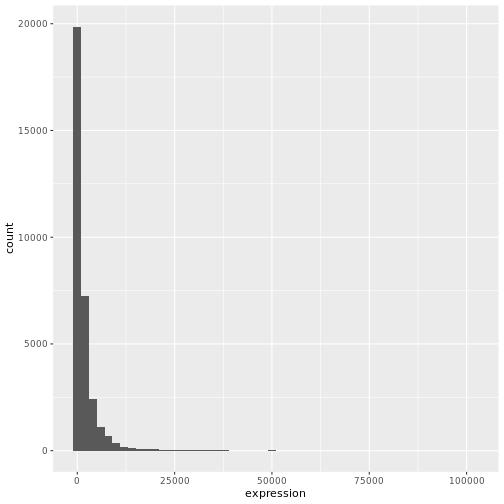
データが右に偏っていることがわかる。 より対称的な分布にするために、
log2変換を適用することができる。 ここでは、
、0に等しい式の値に対して返される-Inf値
を避けるために、小さな定数値(+1)を追加していることに注意してください。
R
rna<- rna %>%
mutate(expression_log = log2(expression + 1))
ここで対数変換した式のヒストグラムを描いてみると、 の分布は確かに正規分布に近くなっている。
R
ggplot(rna, aes(x = expression_log)) + geom_histogram()
出力
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.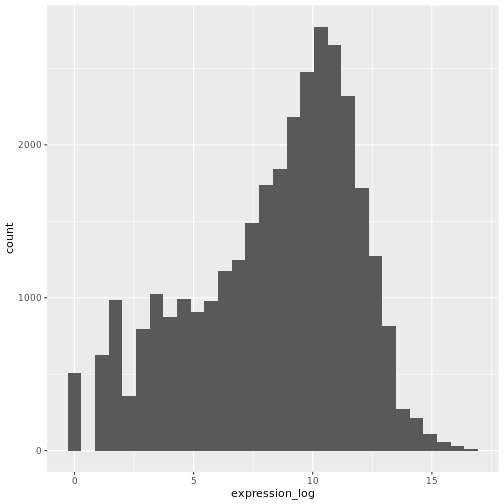
これからは対数変換した発現値を扱うことにする。
R
ggplot(data = rna,mapping = aes(x = expression))+
geom_histogram() +
scale_x_log10()
警告
Warning in scale_x_log10(): log-10 transformation introduced infinite values.出力
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.警告
Warning: Removed 507 rows containing non-finite outside the scale range
(`stat_bin()`).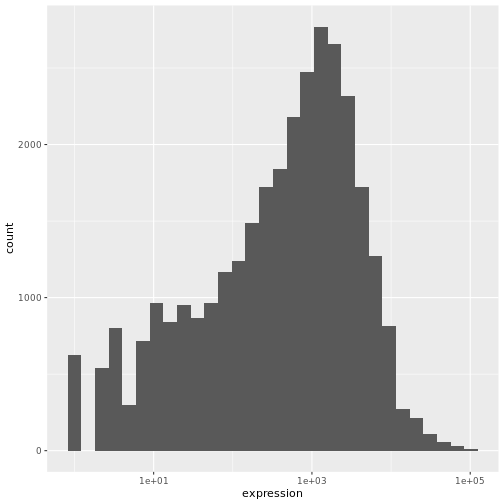
注*。
- ggplot()
関数で設定したものはすべて、あなたが追加したgeom レイヤーで見ることができます(つまり、これらはグローバルなプロット設定です)。 この には、aes()`で設定したx軸とy軸のマッピングが含まれる。 - また、
ggplot()関数でグローバルに定義された マッピングとは別に、与えられたジオムに対するマッピングを指定することもできます。 - 新しいレイヤーを追加するために使われる
+記号は、_前の_レイヤーを含む 行の最後に置かなければなりません。 その代わりに、+記号が 新しいレイヤーを含む行の先頭に追加された場合、ggplot2は新しいレイヤーを追加せず、エラー メッセージを返す。
R
# これはレイヤーを追加するための正しい構文です
rna_plot +
geom_histogram()
# これは新しいレイヤーを追加せず、エラーメッセージを返します
rna_plot
+ geom_histogram()
反復的にプロットを構築する
ここでは、2つの連続変数と
geom_point()関数を使って散布図を描きます。
このグラフは、時間8と時間0、時間4と時間0を比較した発現のlog2倍変化 。
この目的のために、まず対数変換した
発現値の平均値を遺伝子ごと、時間ごとに計算する必要がある。次に、
、時間8と時間0の間の平均対数発現と、
、時間4と時間0の間の平均対数発現を差し引くことにより、対数倍変化を計算する。
ここでは、後で遺伝子を表現するために使用する遺伝子
バイオタイプも含めていることに注意されたい。 フォールドの変化を
rna_fc. という新しいデータフレームに保存する。
R
rna_fc <- rna %>% select(gene, time,
gene_biotype, expression_log) %>%
group_by(gene, time, gene_biotype) %>%
summarize(mean_exp = mean(expression_log)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
mutate(time_8_vs_0 = `8` - `0`, time_4_vs_0 = `4` - `0`)
出力
`summarise()` has grouped output by 'gene', 'time'. You can override using the
`.groups` argument.新しく作成されたデータセット rna_fc を使って ggplot
を作成することができる。
ggplot2`でプロットを作成するのは、通常、反復的なプロセスである。
まず、使用するデータセットを定義し、軸を配置し、 ジオムを選択する:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point()
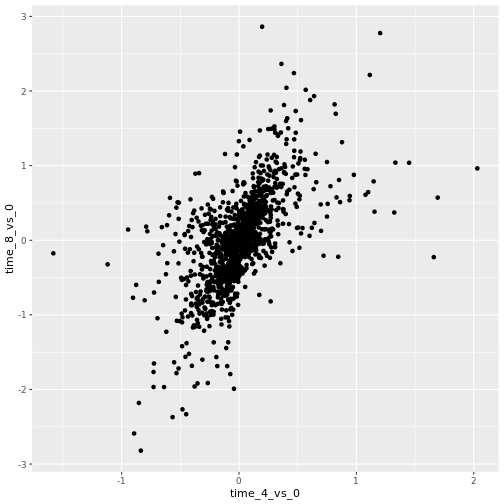
次に、このプロットからより多くの情報を抽出するために、プロットを修正し始める。
例えば、オーバープロットを避けるために透明度(α)を加えることができる:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3)
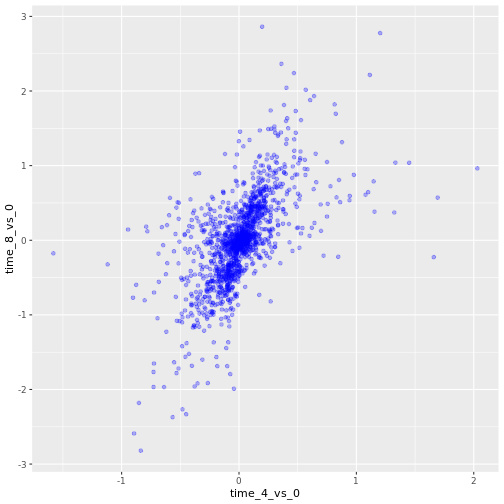
また、すべてのポイントに色を付けることもできる:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3, color = "blue")
あるいは、プロット中の各遺伝子を異なる色にするために、
、引数colorの入力としてベクトルを使うこともできる。
ggplot2は、ベクトルの異なる値に対応する異なる 。 以下は、gene_biotype`を使った例である:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3, aes(color = gene_biotype))

また、
ggplot()関数で提供されるマッピングの中で直接色を指定することもできる。
これはどのジオムレイヤーでも見ることができ、 のマッピングは
aes() で設定したx軸とy軸によって決定される。
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_point(alpha = 0.3)

最後に、geom_abline() :
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_point(alpha = 0.3) +
geom_abline(intercept = 0)
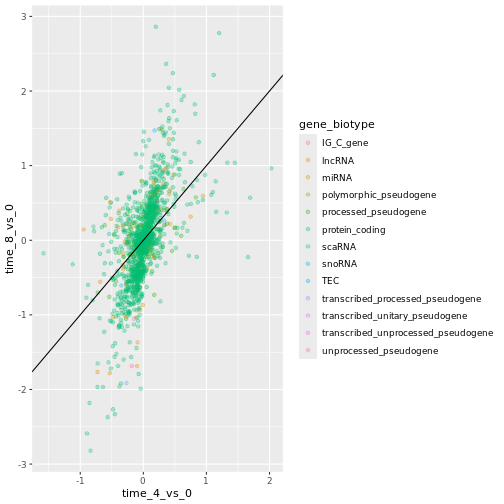
ジオムレイヤーを geom_point から
geom_jitter に変更しても、色は gene_biotype
によって決定されることに注意してください。
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_jitter(alpha = 0.3) +
geom_abline(intercept = 0)

エラー
Error in library("hexbin"): there is no package called 'hexbin'チャレンジ
散布図は、小規模なデータセットの探索に役立つツールである。 rna_fc` データセットのような多数のオブザベーションを持つ データセットの場合、点のオーバープロットは散布図の制限となりうる。 このような設定を扱うための1つの戦略は、 の観測値を六角形にビニングすることである。 プロット空間は六角形にテッセレーションされている。 それぞれの 六角形は、 その境界内に入るオブザベーションの数に基づいて色が割り当てられる。
ggplot2
で六角ビニングを使用するには、まずRパッケージhexbin`をCRANからインストールしてロードする。そして、
geom_hex()関数を使ってhexbin図を作成する。散布図と比較して、六角形のビン プロットの相対的な長所と短所は何か? 上記の散布図( )を調べ、作成した六角形のビンプロットと比較する。
R
install.packages("hexbin")
R
library("hexbin")
エラー
Error in library("hexbin"): there is no package called 'hexbin'R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_hex() +
geom_abline(intercept = 0)
警告
Warning: Computation failed in `stat_binhex()`.
Caused by error in `compute_group()`:
! The package "hexbin" is required for `stat_bin_hex()`.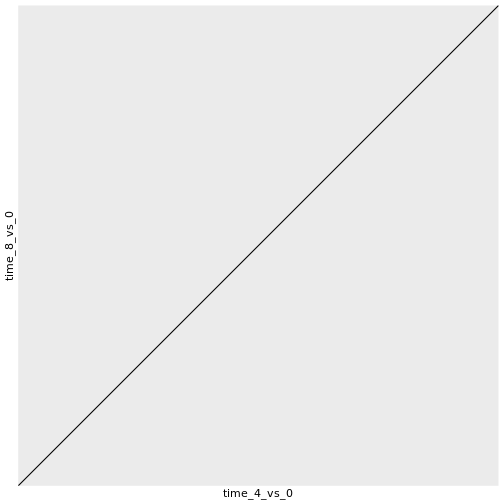
R
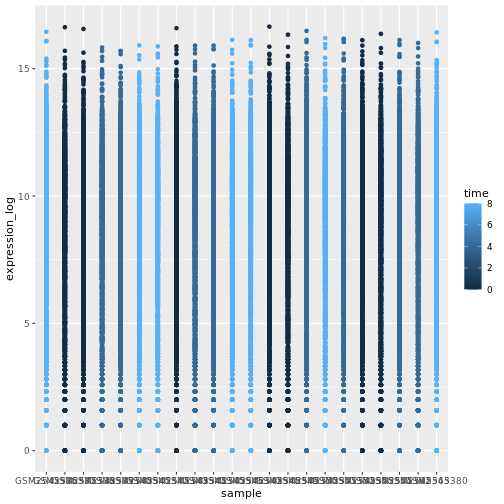
ggplot(data = rna, mapping = aes(y = expression_log, x = sample)) +
geom_point(aes(color = time))
ボックスプロット
ボックスプロットを使って、各サンプル内の遺伝子発現の分布( )を可視化することができる:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_boxplot()

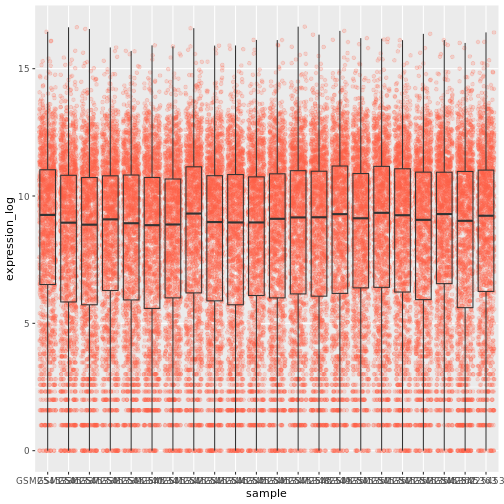
boxplotにポイントを追加することで、 の測定数とその分布をよりよく知ることができる:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_jitter(alpha = 0.2, color = "tomato") +
geom_boxplot(alpha = 0)
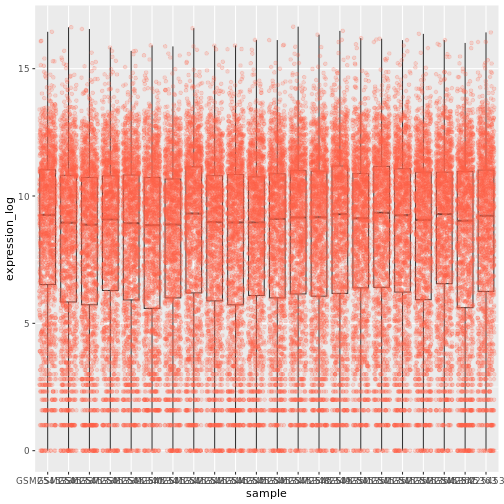
この2つのジオムの順番を入れ替えるべきだ:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample))+
geom_boxplot(alpha = 0) +
geom_jitter(alpha = 0.2, color = "tomato")
X軸の値がまだ正しく 読めないことにお気づきかもしれない。 ラベルの向きを変え、 縦と横に重ならないように調整しよう。 90度の角度を使ってもいいし、 斜め向きのラベルに適切な角度を見つけるために実験してもいい:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample))+
geom_jitter(alpha = 0.2, color = "tomato") +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
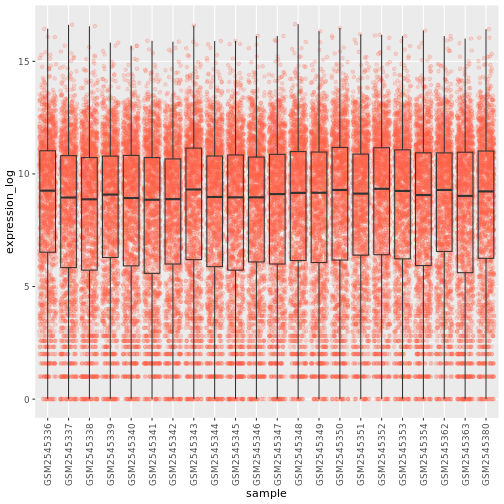
R
# time as integer
ggplot(data = rna,
mapping = aes(y = expression_log,
x = sample))+
geom_jitter(alpha = 0.2, aes(color = time))+
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
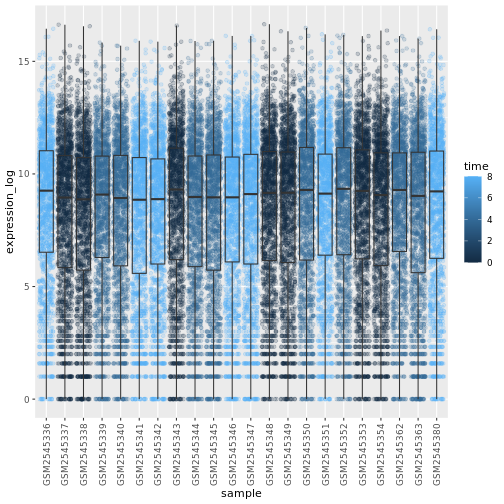
R
# time as factor
ggplot(data = rna,
mapping = aes(y = expression_log,
x = sample))+
geom_jitter(alpha = 0.2, aes(color = as.factor(time)))+
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
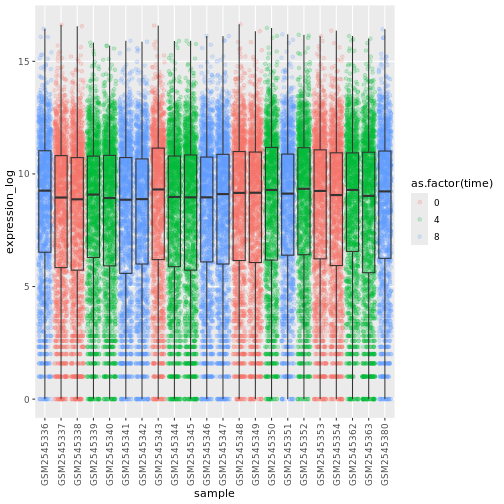
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample))+
geom_violin(aes(fill = as.factor(time)))+
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
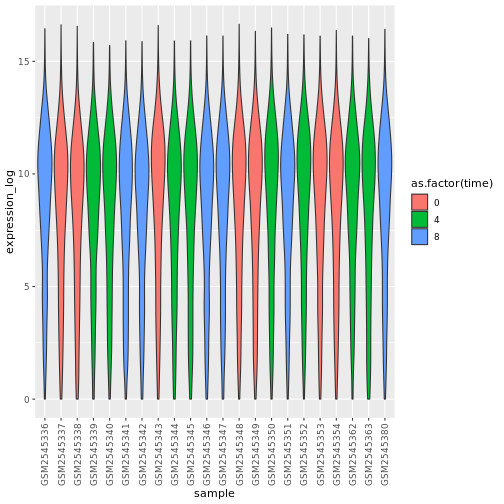
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample))+
geom_violin(aes(fill = sex))+
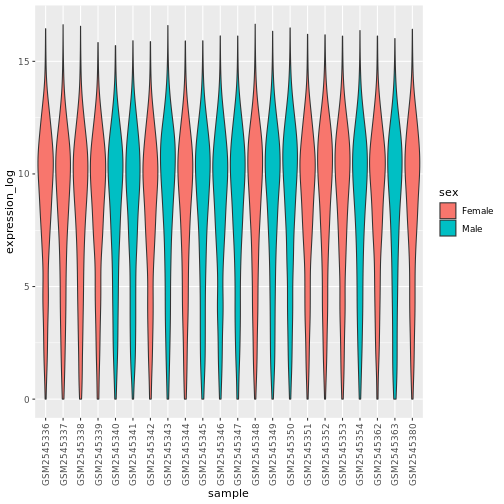
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
線プロット
時刻8と 時刻0を比較し、log
fold変化が最も大きかった10の遺伝子について、感染期間ごとの平均発現量を計算してみよう。
まず、遺伝子を選択し、選択した10遺伝子を含むsub_rnaと呼ばれるrna
のサブセットを作成する必要がある。次に、
データをグループ化し、各グループ内の平均遺伝子発現を計算する必要がある:
R
rna_fc<- rna_fc %>% arrange(desc(time_8_vs_0))
genes_selected <- rna_fc$gene[1:10]
sub_rna<- rna %>%
filter(gene %in% genes_selected)
mean_exp_by_time<- sub_rna %>%
group_by(gene,time) %>%
summarize(mean_exp = mean(expression_log))
出力
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.R
mean_exp_by_time
出力
# A tibble: 30 × 3
# Groups: gene [10]
gene time mean_exp
<chr> <int> <dbl>
1 Acr 0 5.07
2 Acr 4 5.54
3 Acr 8 7.31
4 Aipl1 0 3.70
5 Aipl1 4 3.89
6 Aipl1 8 6.56
7 Bst1 0 3.20
8 Bst1 4 3.77
9 Bst1 8 5.22
10 Chil3 0 4.00
# ℹ 20 more rowsX軸に感染期間( )、Y軸に平均発現をとって折れ線グラフを作成することができる:
R
ggplot(data = mean_exp_by_time, mapping = aes(x = time, y = mean_exp))+
geom_line()
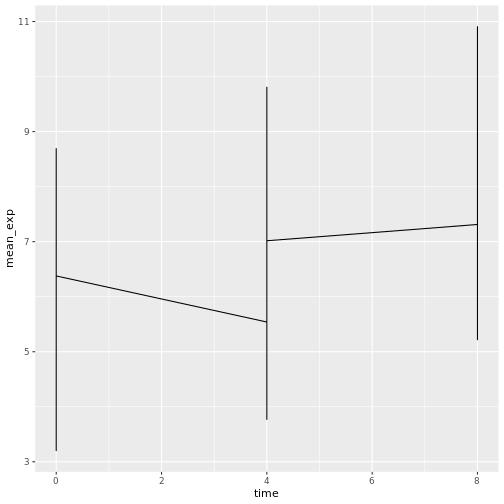
残念なことに、これはうまくいかない。というのも、
の全遺伝子のデータをまとめてプロットしたからである。
各遺伝子に対して線を引くようにggplotに指示する必要がある。
、group = geneを含むようにesthetic関数を修正する:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp, group = gene))+
geom_line()
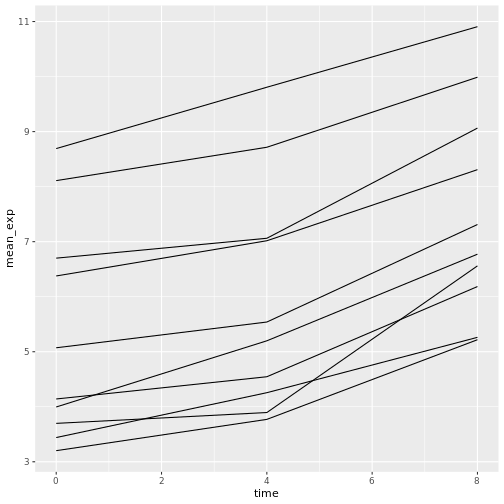
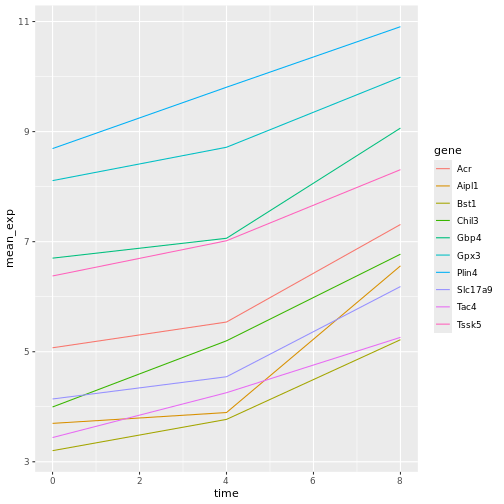
色をつければ、プロットの中で遺伝子を区別できるようになる(
colorを使えば、自動的にデータをグループ化することもできる):
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp, color = gene))+
geom_line()
ファセット
ggplot2`には_faceting_と呼ばれる特別なテクニックがあり、 、データセットに含まれる 、1つのプロットを複数の(サブ)プロットに分割することができる。 こ れ ら の異な る サブプ ロ ッ ト は、 同 じ プ ロ パテ ィ を継承 し ます (軸の限界、目盛り、 …)。 直接比較しやすくするためだ。 、これを用いて各遺伝子について時間軸に沿った折れ線グラフを作成する:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp))+ geom_line() +
facet_wrap(~ gene)
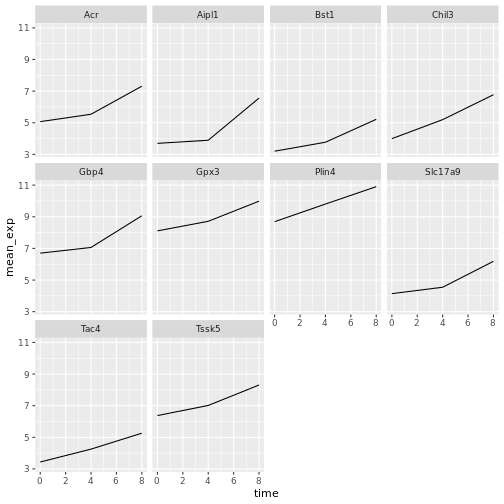
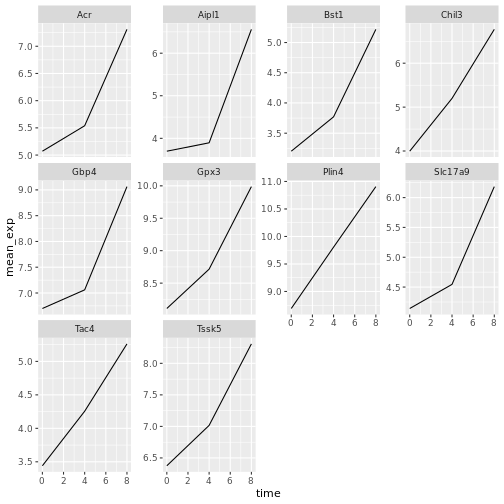
ここでは、X軸とY軸はすべてのサブプロットで同じスケールになっている。、Y軸のスケールを自由に設定できるようにscalesを修正することで、このデフォルトの動作を変更することができる:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp))+
geom_line() +
facet_wrap(~ gene, scales = "free_y")
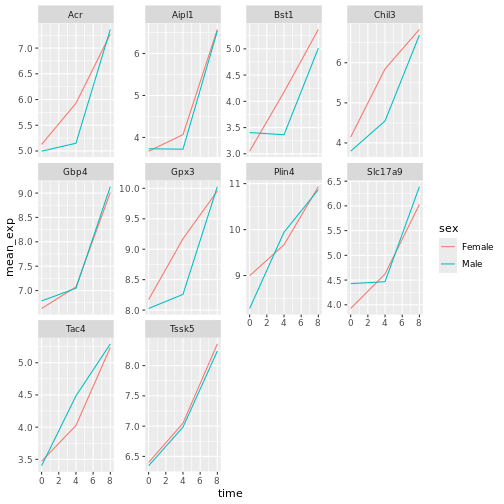
ここで、各プロットの線をマウスの性別で分けたい。 そのためには、 、遺伝子、時間、性別でグループ化したデータフレームの平均発現を計算する必要がある:
R
mean_exp_by_time_sex<- sub_rna %>%
group_by(gene, time, sex) %>%
summarize(mean_exp = mean(expression_log))
出力
`summarise()` has grouped output by 'gene', 'time'. You can override using the
`.groups` argument.R
mean_exp_by_time_sex
出力
# A tibble: 60 × 4
# Groups: gene, time [30]
gene time sex mean_exp
<chr> <int> <chr> <dbl>
1 Acr 0 Female 5.13
2 Acr 0 Male 5.00
3 Acr 4 Female 5.93
4 Acr 4 Male 5.15
5 Acr 8 Female 7.27
6 Acr 8 Male 7.36
7 Aipl1 0 Female 3.67
8 Aipl1 0 Male 3.73
9 Aipl1 4 Female 4.07
10 Aipl1 4 Male 3.72
# ℹ 50 more rowscolor
を使ってさらに分割することで、ファセット化されたプロットを作ることができる(1つのプロット内で):
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex))+
geom_line() +
facet_wrap(~ gene, scales = "free_y")
通常、背景が白いプロットは印刷したときに読みやすくなる。 、関数
theme_bw() を使って背景を白に設定することができる。
さらに、グリッドを削除することもできる:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex))+
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
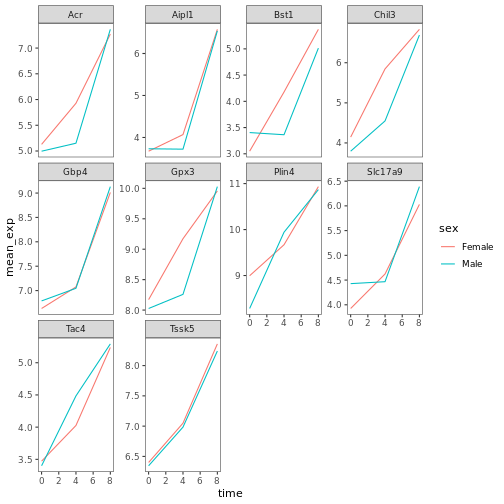
R
mean_exp_by_chromosome<- rna %>%
group_by(chromosome_name, time) %>%
summary(mean_exp = mean(expression_log))
ggplot(data = mean_exp_by_chromosome, mapping = aes(x = time,
y = mean_exp))+
geom_line() +
facet_wrap(~ chromosome_name, scales = "free_y")
エラー
Error in `fortify()`:
! `data` must be a <data.frame>, or an object coercible by `fortify()`,
or a valid <data.frame>-like object coercible by `as.data.frame()`.
Caused by error in `.postvalidate_data_frame_like_object()`:
! `as.data.frame(data)` must preserve dimensions.facet_wrapジオメトリは、1ページにきれいに収まるように、プロットを任意の数の 次元に抽出する。 一方、facet_gridジオメトリでは、 数式表記(rows
~ columns;.`
は、1つの行または列のみを示すプレースホルダとして使用できます)によって、プロットの配置方法を明示的に指定することができます。
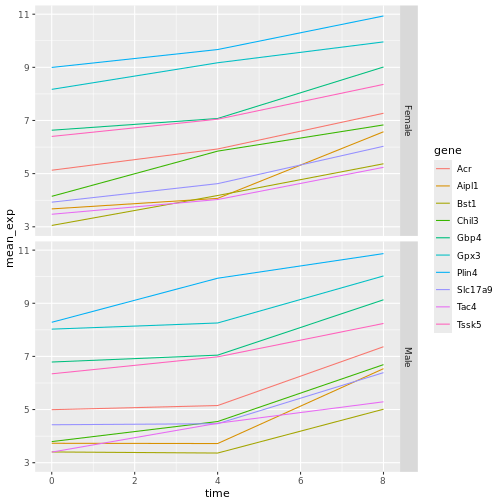
先ほどのプロットを修正し、男性と女性の平均遺伝子発現 が経時的にどのように変化したかを比較してみよう:
R
# 1列、行ごとのファセット
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = gene))+
geom_line() +
facet_grid(sex ~ .)
R
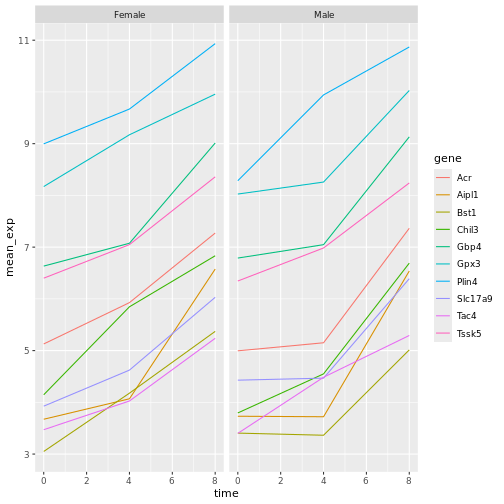
# 1行、列ごとのファセット
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = gene))+
geom_line() +
facet_grid(. ~ sex)
ggplot2`テーマ
ggplot2 には、プロットの背景を白に変更する
theme_bw() に加えて、
手早く視覚化の見た目を変更するのに便利なテーマがいくつか用意されている。
テーマの全リストはhttps://ggplot2.tidyverse.org/reference/ggtheme.htmlで入手できる。
theme_minimal()やtheme_light()は人気があり、theme_void()`
は新しい手作りのテーマを作る出発点として役に立つ。
ggthemes](https://jrnold.github.io/ggthemes/reference/index.html)
パッケージは様々なオプション(Excel 2003 テーマを含む)を提供します。
ggplot2 extensions website](https://exts.ggplot2.tidyverse.org/) は、追加の
テーマを含む ggplot2 の機能を拡張する
パッケージのリストを提供しています。
カスタマイズ
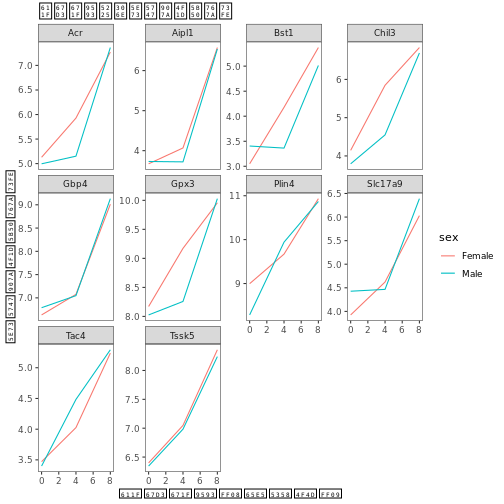
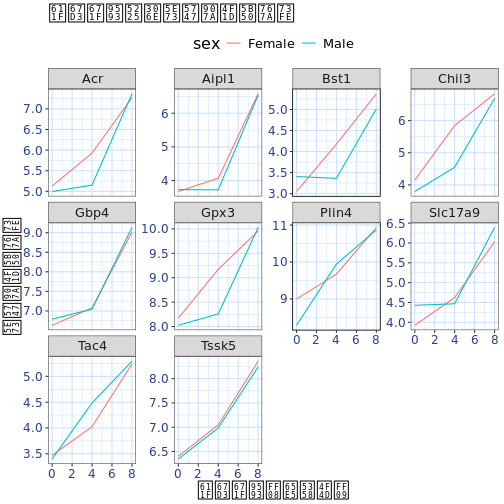
時間別、遺伝子別の平均発現のファセット・プロットに戻ろう。 性別に色分けされている。
ggplot2`のカンニング シート](https://raw.githubusercontent.com/rstudio/cheatsheets/main/data-visualization.pdf), を見て、プロットを改善する方法を考えてみてください。
ここで、軸の名前を ‘time’ や ‘mean_exp’ よりも情報量の多いものに変更し、図にタイトルを追加する:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex))+
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "感染期間別の平均遺伝子発現",
x = "感染期間(日単位)",
y = "平均遺伝子発現")
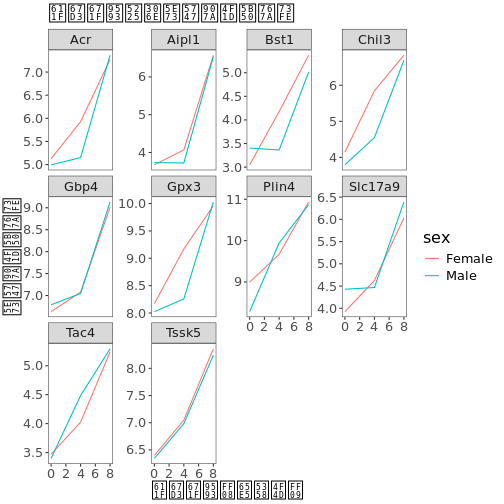
軸にはより情報量の多い名前が付けられているが、フォントサイズを大きくすることで読みやすさは :
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex))+
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "感染期間別の平均遺伝子発現",
x = "感染期間(日単位)",
y = "平均遺伝子発現") +
theme(text = element_text(size = 16))
プロットのフォントを変更することも可能です。 Windowsをお使いの場合は、 をインストールする必要があるかもしれません。
さらに、X軸とY軸のテキストの色、
グリッドの色などをカスタマイズできる。 例えば、
legend.positionを"top"に設定することで、凡例を一番上に移動させることもできる。
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex))+
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "感染期間別の平均遺伝子発現",
x = "感染期間(日単位)",
y = "平均遺伝子発現") +
theme(text = element_text(size = 16),
axis.text.x = element_text(color = "royalblue4", size = 12),
axis.text.y = element_text(color = "royalblue4", size = 12),
panel.grid = element_line(color="lightsteelblue1"),
legend.position = "top")
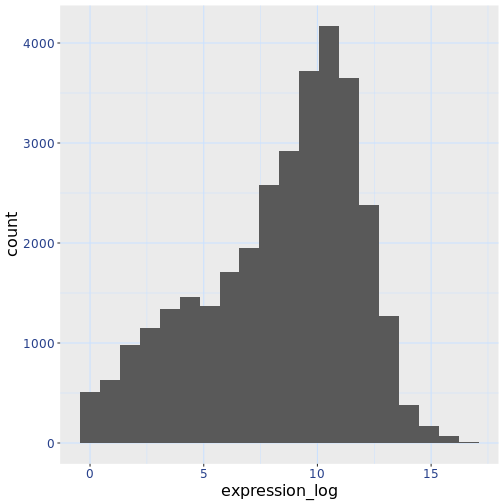
作成した変更がデフォルトのテーマよりも気に入った場合は、 、オブジェクトとして保存して、作成した他の プロットに簡単に適用することができます。 以下は、 以前に作成したヒストグラムを使った例です。
R
blue_theme <- theme(axis.text.x = element_text(colour = "royalblue4",
size = 12),
axis.text.y = element_text(colour = "royalblue4",
size = 12),
text = element_text(size = 16),
panel.grid = element_line(colour="lightsteelblue1"))
ggplot(rna, aes(x = expression_log)) +
geom_histogram(bins = 20) +
blue_theme
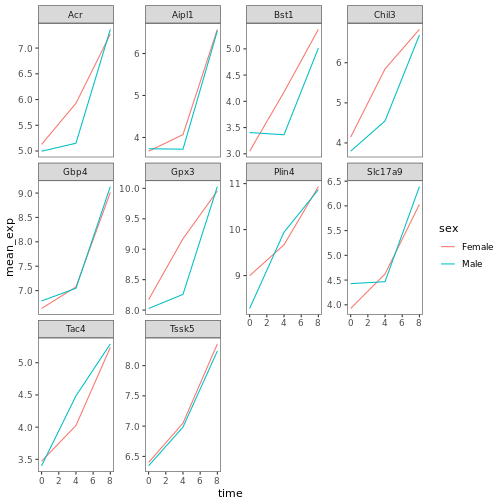
チャレンジ
これらの情報を手に入れたら、 、この練習で作成したプロットのいずれかを改良するか、 、あなた自身の美しいグラフを作成してください。 RStudio ggplot2 を参考にしてください。 いくつかアイデアを挙げてみよう:
- 線の太さを変えてみてください。
- 伝説の名前を変える方法はありますか?
そのラベルについてはどうだろう? (ヒント:
scale_で始まる ggplot 関数を探す) - 別のカラーパレットを使用するか、線の カラーを手動で指定してみてください( http://www.cookbook-r.com/Graphs/Colors_(ggplot2)/ 参照)。
例えば、このプロットに基づくと
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex))+
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
以下のようなカスタマイズが可能です:
R
# change the thickness of the lines
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line(size=1.5) +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
警告
Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use `linewidth` instead.
This warning is displayed once every 8 hours.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.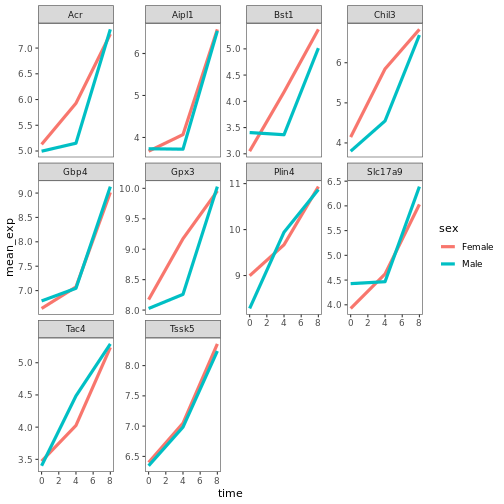
R
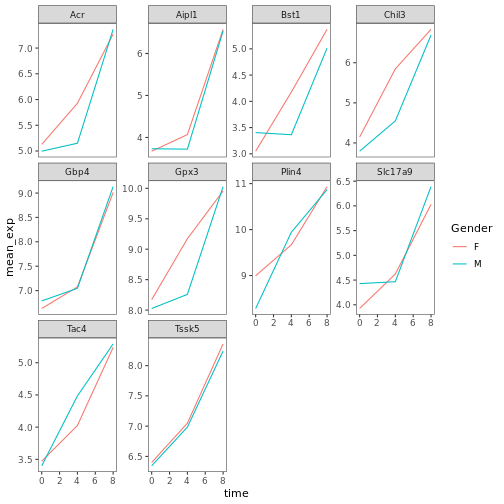
# change the name of the legend and the labels
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_discrete(name = "Gender", labels = c("F", "M"))
R
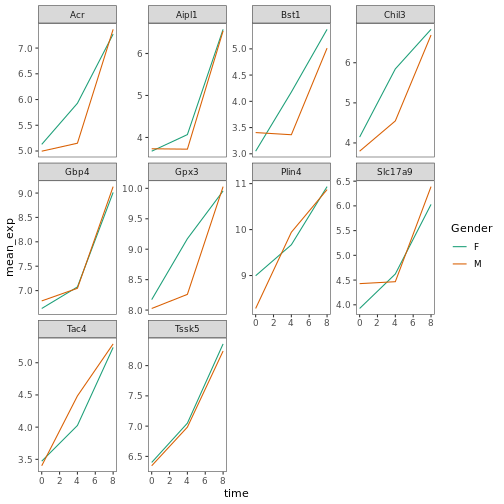
# using a different color palette
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_brewer(name = "Gender", labels = c("F", "M"), palette = "Dark2")
R
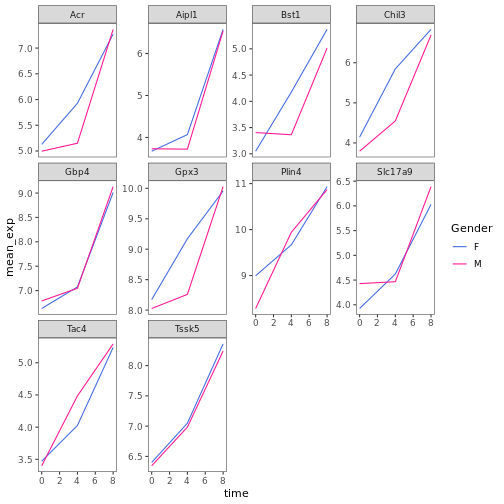
# manually specifying the colors
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_manual(name = "Gender", labels = c("F", "M"),
values = c("royalblue", "deeppink"))
プロットの構成
ファセッティングは、1つのプロットを複数のサブプロットに分割するのに最適なツールです。 しかし、 複数の独立したプロット、すなわち異なる 変数、あるいは異なるデータフレームに基づくプロットを含む1つの図を作成したい場合があります。
、まず2つのプロットを作成する:
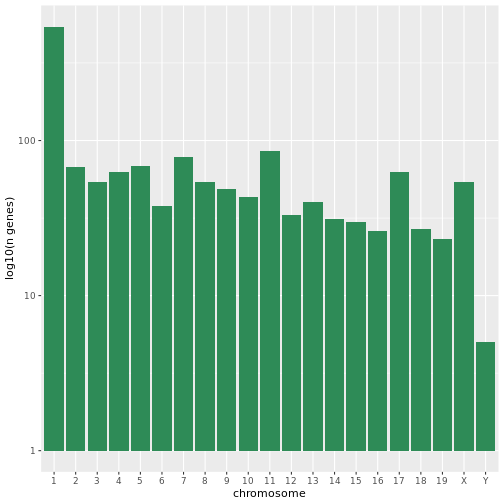
最初のグラフは、染色体ごとにユニークな遺伝子の数を数えている。
、まずchromosome_nameのレベルを並べ替え、
、染色体ごとにユニークな遺伝子をフィルタリングする必要がある。
また、読みやすくするために、Y軸のスケールを
log10スケールに変更した。
R
rna$chromosome_name <- factor(rna$chromosome_name,
levels = c(1:19,"X","Y"))
count_gene_chromosome <- rna %>% select(chromosome_name, gene) %>%
distinct() %>% ggplot() +
geom_bar(aes(x = chromosome_name), fill = "seagreen",
position = "dodge", stat = "count") +
labs(y = "log10(n genes)", x = "chromosome") +
scale_y_log10()
count_gene_chromosome
以下では、 legend.position を "none"
に設定することで、凡例を完全に削除することもできる。
R
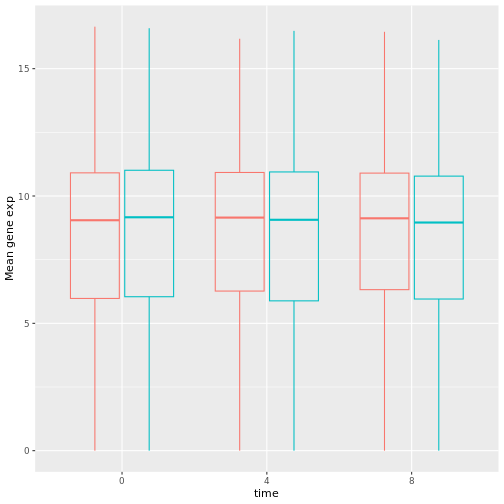
exp_boxplot_sex <- ggplot(rna, aes(y=expression_log, x = as.factor(time),
color=sex)) +
geom_boxplot(alpha = 0) +
labs(y = "Mean gene exp",
x = "time") + theme(legend.position = "none")
exp_boxplot_sex
patchwork**](https://github.com/thomasp85/patchwork)パッケージ
は、+
を使って図形を組み合わせるエレガントなアプローチを提供し、
図形を(通常は横に並べて)並べます。 より具体的には、|
は明示的に横に並べ、/は 上に重ねる。
R
install.packages("patchwork")
R
library("patchwork")
エラー
Error in library("patchwork"): there is no package called 'patchwork'R
count_gene_chromosome + exp_boxplot_sex
エラー
Error in `ggplot_add()`:
! Can't add `exp_boxplot_sex` to a <ggplot> object.R
## or count_gene_chromosome | exp_boxplot_sex
R
count_gene_chromosome / exp_boxplot_sex
エラー
Error in count_gene_chromosome/exp_boxplot_sex: non-numeric argument to binary operatorplot_layout
と組み合わせることで、最終的なコンポジションのレイアウトをさらにコントロールし、より複雑なレイアウトを作成することができる:
R
count_gene_chromosome + exp_boxplot_sex + plot_layout(ncol = 1)
エラー
Error in `ggplot_add()`:
! Can't add `exp_boxplot_sex` to a <ggplot> object.R
count_gene_chromosome +
(count_gene_chromosome + exp_boxplot_sex) +
exp_boxplot_sex +
plot_layout(ncol = 1)
エラー
Error in `ggplot_add()`:
! Can't add `exp_boxplot_sex` to a <ggplot> object.最後のプロットは | と /
のコンポーザーを使っても作成できる:
R
count_gene_chromosome /
(count_gene_chromosome | exp_boxplot_sex) /
exp_boxplot_sex
エラー
Error in count_gene_chromosome | exp_boxplot_sex: operations are possible only for numeric, logical or complex typesパッチワークの詳細については、 ウェブページ、またはこちらの ビデオをご覧ください。
もう一つのオプションは
**gridExtra** パッケージで、 の別々の ggplot をgrid.arrange()`
を使って一つの図にまとめることができます:
R
install.packages("gridExtra")
R
library("gridExtra")
エラー
Error in library("gridExtra"): there is no package called 'gridExtra'R
grid.arrange(count_gene_chromosome, exp_boxplot_sex, ncol = 2)
エラー
Error in grid.arrange(count_gene_chromosome, exp_boxplot_sex, ncol = 2): could not find function "grid.arrange"引数 ncol と nrow は単純な
の配置を作るのに使われるが、それに加えて、より複雑な のレイアウトを作る
ためのツールもある。
プロットのエクスポート
プロットを作成したら、お好きな 形式でファイルに保存できます。 RStudioのPlotペインのExportタブでは、 のプロットが低解像度で保存されます。これは多くのジャーナルでは受け入れられませんし、 ではポスター用にうまく拡大縮小できません。
代わりに ggsave() 関数を使います。この関数を使うと、
の適切な引数(width, height,
dpi)を調整することで、プロットの
次元と解像度を簡単に変更することができます。
作業ディレクトリに fig_output/
フォルダがあることを確認してください。
R
my_plot <- ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
labs(title = "Mean gene expression by duration of the infection",
x = "Duration of the infection (in days)",
y = "Mean gene expression") +
guides(color=guide_legend(title="Gender")) +
theme_bw() +
theme(axis.text.x = element_text(colour = "royalblue4", size = 12),
axis.text.y = element_text(colour = "royalblue4", size = 12),
text = element_text(size = 16),
panel.grid = element_line(colour="lightsteelblue1"),
legend.position = "top")
ggsave("fig_output/mean_exp_by_time_sex.png", my_plot, width = 15,
height = 10)
# This also works for grid.arrange() plots
combo_plot <- grid.arrange(count_gene_chromosome, exp_boxplot_sex,
ncol = 2, widths = c(4, 6))
ggsave("fig_output/combo_plot_chromosome_sex.png", combo_plot,
width = 10, dpi = 300)
注意: パラメータ width と height
は、保存されたプロットのフォントサイズ も決定します。
その他のビジュアライゼーション用パッケージ
ggplot2`は非常に強力なパッケージで、我々の_tidy data_と_tidy tools_パイプラインにうまくフィットする。 Rには他にも無視できない可視化パッケージ がある。
ベースグラフィック
Rに付属するデフォルトのグラフィックス・システムは、しばしば_ベースR
グラフィックス_と呼ばれ、シンプルで高速である。
これは_画家またはキャンバスの
モデル_に基づいており、異なる出力がそれぞれ 互いに直接重ね合わされる(図@ref(fig:paintermodel)を参照)。
これはggplot2(および後述するlattice)との基本的な
の違いである。
は専用のオブジェクトを返し、それは画面上またはファイル上にレンダリングされ、
は更新することさえできる。
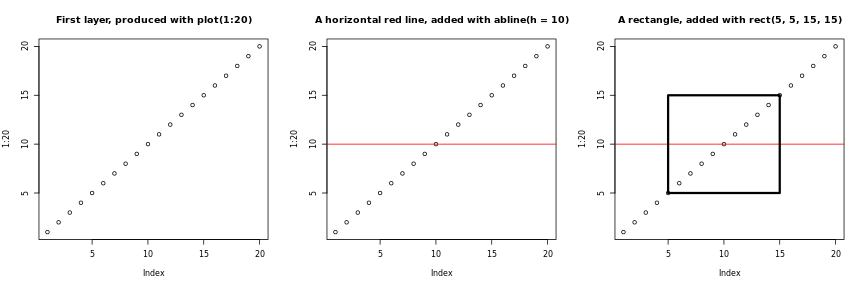
R
par(mfrow = c(1, 3))
plot(1:20, main = "First layer, produced with plot(1:20)")
plot(1:20, main = "A horizontal red line, added with abline(h = 10)")
abline(h = 10, col = "red")
plot(1:20, main = "A rectangle, added with rect(5, 5, 15, 15)")
abline(h = 10, col = "red")
rect(5, 5, 15, 15, lwd = 3)
もうひとつの主な違いは、ベース・グラフィックスのプロット関数は、入力タイプに基づいて、
、_正しい_ことをしようとする。 これは、
データフレームしか入力として受け付けないggplot2で、プロットをビットごとに構築する必要があるのとは、
異なる。
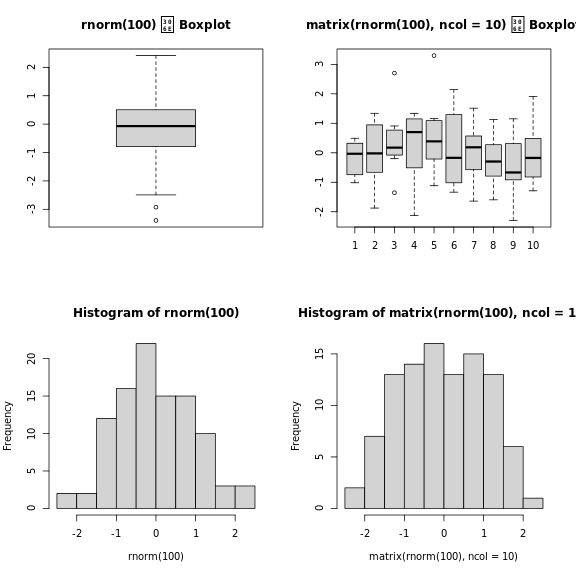
R
par(mfrow = c(2, 2))
boxplot(rnorm(100),
main = "rnorm(100) の Boxplot")
boxplot(matrix(rnorm(100), ncol = 10),
main = "matrix(rnorm(100), ncol = 10) の Boxplot")
hist(rnorm(100))
hist(matrix(rnorm(100), ncol = 10))
、plot、hist、
boxplot、…のような1行のコードと1つの関数で非常に素早く作成できる。
しかし、デフォルトは必ずしも最も魅力的なものではありません。
、図形のチューニングは、特に複雑になると(例えばファセットを生成するために
)、時間がかかり面倒になります。
格子パッケージ
lattice** パッケージはggplot2と似ているが、 データフレームを入力として使い、グラフィカルオブジェクトを返し、ファセットをサポートする。 しかし、lattice`はグラフィックの文法に基づいておらず、
より複雑なインターフェイスを持っている。
lattice`パッケージの良いリファレンスは@latticebookだ。